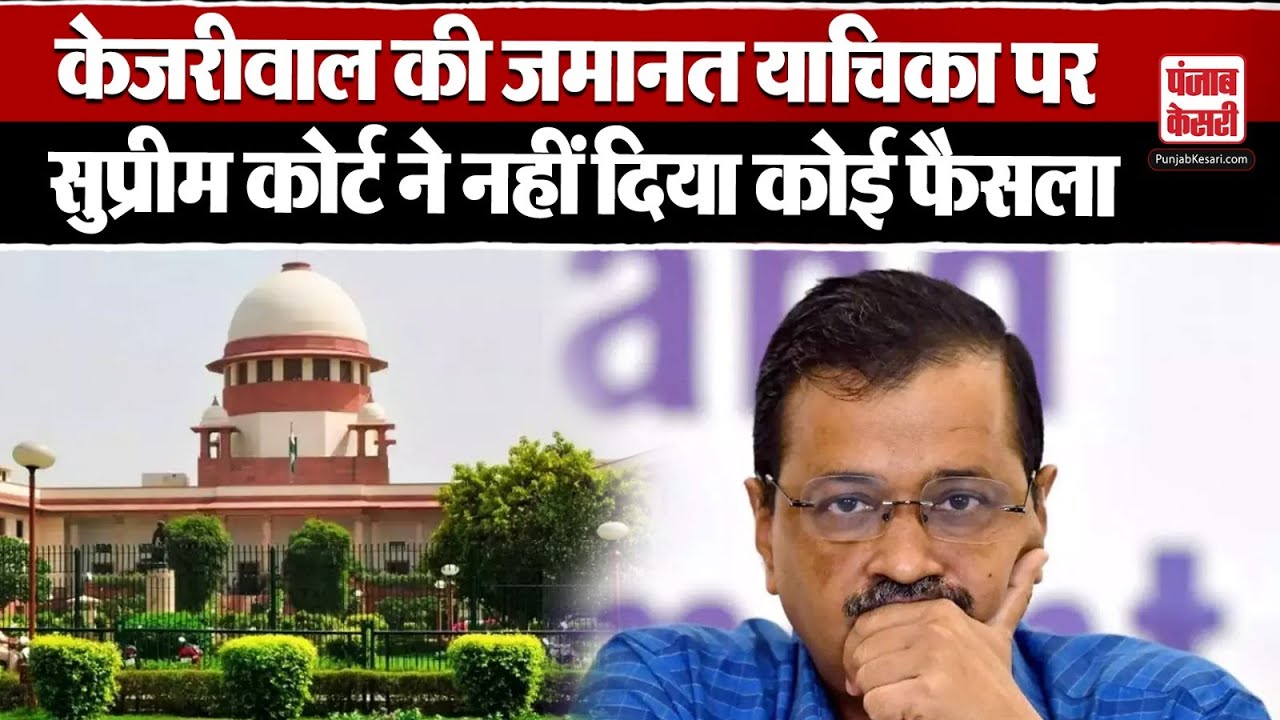




















Lok Sabha Election: मतदान केंद्र के बाहर भारी भीड़ के बीच बुजुर्ग महिला ने PM मोदी को बांधी राखी
Lok Sabha Election: प्रधानमंत्री नरेंद्र मोदी ने मंगलवार को अहमदाबाद के रानिप स्थित निशान स्कूल में वोट डाला। इस दौरान उत्साही भीड़ ने उनका जोरदार

Lok Sabha election: बिहार में पहले दो घंटे में 10.78 प्रतिशत मतदान दर्ज
Lok Sabha election: बिहार में लोकसभा चुनाव के तीसरे चरण में पांच संसदीय क्षेत्र झंझारपुर, अररिया, सुपौल,
Lok Sabha election: छत्तीसगढ़ में पहले दो घंटे में 13 फीसदी से अधिक मतदान दर्ज
Lok Sabha election: लोकसभा चुनाव के तीसरे चरण में छत्तीसगढ़ की शेष सात सीटों पर मंगलवार को

Lok Sabha Election 2024: तीसरे चरण का मतदान शुरू, 9 बजे तक 10.51% डाले गए वोट
Lok Sabha Election 2024: लोकसभा चुनाव 2024 (Lok Sabha Election 2024) के लिए तीसरे चरण का मतदान
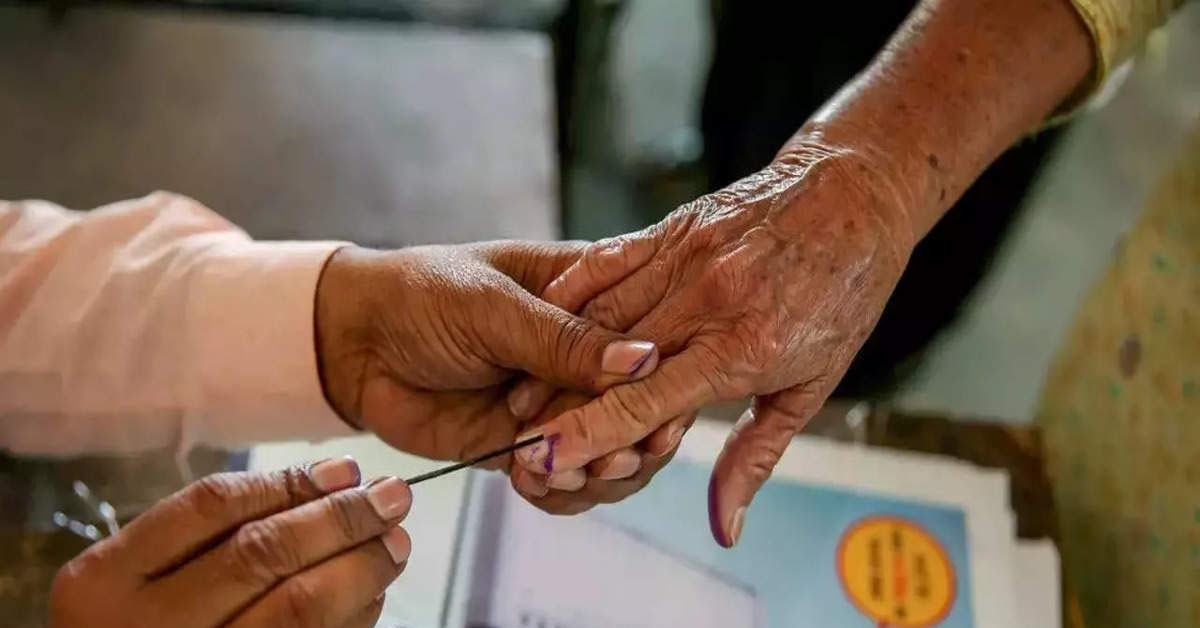
Lok Sabha Election 2024: तीसरे चरण में दोपहर 3 बजे तक 50.71% मतदान दर्ज
Lok Sabha Election 2024: लोकसभा चुनाव 2024 (Lok Sabha Election 2024) के तीसरे चरण के मतदान चल

Lok Sabha Election 2024: दूसरे चरण में 1 बजे तक के अलग-अलग राज्यों में मतदान के ये हैं जारी आंकड़े
Loksabha Election 2024: लोकसभा चुनाव में दूसरे चरण के लिए वोटिंग जारी है। अब तक यानी 1

बंगाल में वोटिंग के दौरान हिंसा की घटना, BJP ने लगाया TMC पर आरोप
West Bengal Violence: पश्चिम बंगाल के भाजपा प्रमुख और बालुरघाट लोकसभा सीट के उम्मीदवार सुकांत मजूमदार और
राजस्थान की 13 लोकसभा सीटों पर पिछले दो घंटे में 11 प्रतिशत से अधिक मतदान
Rajasthan Lok Sabha: राजस्थान की 13 लोकसभा सीटों पर शुक्रवार को सुबह सात बजे मतदान शुरू हुआ और
Lok Sabha Election 2024: तीसरे चरण में सुबह 11 बजे तक 25.41% मतदान दर्ज
Lok Sabha Election 2024: लोकसभा चुनाव 2024 (Lok Sabha Election 2024) के लिए तीसरे चरण का मतदान
Lok Sabha Election 2024: बंगाल में तीसरे चरण के मतदान में जारी हिंसा, कई मतदान केंद्रों से आई खबरें
Lok Sabha Election 2024: लोकसभा चुनाव 2024 (Lok Sabha Election 2024) के तीसरे चरण के मतदान के
देश की सबसे हॉट सीट बाड़मेर-जैसलमेर में मतदान शुरू, कैलाश चौधरी ने डाला वोट
Jaisalmer Voting: देश की सबसे हॉट सीट बाड़मेर-जैसलमेर में सुबह सात बजे से मतदान शुरू हो गया है।

Elon Musk का दौरा रद्द होने पर कांग्रेस पार्टी का सरकार पर तंज
Delhi: कांग्रेस ने ‘टेस्ला’ के प्रमुख एलन मस्क का भारत दौरा स्थगित होने को लेकर शनिवार को